Computational methods
Long reads
The Shasta assembler can be used to assemble DNA sequence from long reads. It is optimized especially for Oxford Nanopore reads, but can also be used to assemble other types of long reads such as those generated by Pacific Biosciences sequencing platforms.
Oxford Nanopore reads are rapidly evolving in characteristics, and therefore cannot be characterized precisely. Here is a summary of read metrics for the best available reads as of September 2020:
- Typical length around 50 Kb, with just a few percent of coverage in reads below 10 Kb and 10% or more of coverage in reads longer than 100 Kb. There are also Ultra-Long (UL) protocols that provide longer reads, with typical lengths around 100 Kb.
- Sequence identity around 97%, or equivalently an error rate around 3%. This relatively high error rate has been improving significantly in time.
- The dominant error mode consists of errors in the length of homopolymer runs of all lengths. Errors in short homopolymer runs (lengths 1-5) are particularly deleterious due to their high frequency.
Computational challenges
Due to their length, Oxford Nanopore reads have unique value for de novo assembly. However, a successful approach needs to deal with the high error rate.
Traditional approaches to de novo assembly typically rely on selecting a k-mer length that satisfies the following:
- K-mers of the selected length are reasonably unique in the target genome. For human assemblies, this typically means k ⪆ 30.
- Most k-mers of the selected length are error-free in the target reads. This means that k must be significantly less than the inverse of the error rate.
For many sequencing technolgies, the above conditions are satisfied for k around 30. But for our target reads, with an error rate around 3%, most 30-mers contain errors, and therefore assembly algorithms based on using such k-mers become unfeasible.
A possible approach consists of adding a preliminary error correction step in which reads are aligned to each other and corrected based on consensus and then presenting to the assembler the corrected reads, which hopefully have a much lower error rate. But such approaches tend to be slow, and in addition, any errors made in the error correction step are permanent and cannot be fixed during the assembly process.
Read representation
The computational techniques used in the Shasta assembler rely on representing the sequence of input reads in a way that reduces the effect of errors on the de novo assembly process:
- The sequence of input reads is represented using run-length encoding.
- In many assembly steps, the sequence of input reads is described by using occurrences of a pre-determined subset of short k-mers (k ≈ 10 in run-encoding) called markers.
Run-length encoding
With run-length encoding, the sequence of each input read is represented as a sequence of bases, each with a repeat count that says how many times each of the bases is repeated. For example, the following read
CGATTTAAGTTAis represented as follows using run-length encoding:
CGATAGTA 11132121
Using run-length encoding makes the assembly process less sensitive to errors in the length of homopolymer runs, which are the most common type of errors in Oxford Nanopore reads. For example, consider these two reads:
CGATTTAAGTTA CGATTAAGGGTTAUsing their raw representation above, these reads can be aligned like this:
CGATTTAAG--TTA CGATT-AAGGGTTAAligning the second read to the first required a deletion and two insertions. But in run-length encoding, the two reads become:
CGATAGTA 11132121 CGATAGTA 11122321The sequence portions are now identical and can be aligned trivially and exactly, without any insertions or deletions:
CGATAGTA CGATAGTAThe differences between the two reads only appear in the repeat counts:
11132121 11122321 * *
The Shasta assembler uses one byte to represent repeat counts, and as a result, it only represents repeat counts between 1 and 255. If a read contains more than 255 consecutive bases, it is discarded on input. Such reads are extremely rare, and the occurrence of such a large number of repeated bases is probably a symptom that something is wrong with the read anyway.
Some properties of base sequences in run-length encoding
- In the sequence portion of the run-length encoding, consecutive bases are always distinct. If they were not, the second one would be removed from the run-length encoded sequence, while increasing the repeat count for the first one.
- With ordinary base sequences, the number of distinct k-mers of length k is 4k. But with run-length base sequences, the number of distinct k-mers of length k is 4×3k-1. This is a consequence of the previous bullet.
- The run-length sequence is generally shorter than the raw sequence, and cannot be longer. For a long random sequence, the number of bases in the run-length representation is 3/4 of the number of bases in the raw representation.
Markers
Even with run-length encoding, errors in input reads are still frequent. To further reduce sensitivity to errors, and also to speed up some of the computational steps in the assembly process, the Shasta assembler also uses a read representation based on markers. Markers are occurrences in reads of a pre-determined subset of short k-mers. By default, Shasta uses for this purpose k-mers with k=10 in run-length encoding, corresponding to an average of approximately 13 bases in raw read representation.
Just for illustration, consider a description using markers of length 3 in run-length encoding. There is a total 4×32 = 36 distinct such markers. We arbitrarily choose the following fixed subset of the 36, and we assign an id to each of the kmers in the subset:
TGC | 0 |
GCA | 1 |
GAC | 2 |
CGC | 3 |
Consider now the following portion of a read in run-length representation (here, the repeat counts are irrelevant and so they are omitted):
CGACACGTATGCGCACGCTGCGCTCTGCAGC
GAC TGC CGC TGC
CGC TGC GCA
GCA CGC
Occurrences of the k-mers defined in the table above are shown
and define the markers in this read. Note that markers
can overlap. Using the marker ids defined in the table above,
we can summarize the sequence of this read portion as follows:
2 0 3 1 3 0 3 0 1This is the marker representation of the read portion above. It just includes the sequence of markers occurring in the read, not their positions.
In Shasta documentation and code, the zero-based sequence number of a marker in the marker representation of an oriented read is called the marker ordinal or simply ordinal. The first marker in an oriented read has ordinal 0, the second one has ordinal 1, and so on.
Note that the marker representation loses information, as it is not possible to reconstruct the complete initial sequence from the marker representation. This also means that the marker representation is insensitive to errors in the sequence portions that don't belong to any markers.
The Shasta assembler uses a random choice of the k-mers to be used
as markers. The length of the markers k is controlled by
assembly parameter
--Kmers.k
with a default value of 10.
Each k-mer is randomly chosen to be used as a marker
with probability determined by assembly parameter
--Kmers.probability
with a default value of 0.1.
There are 4 possibilities for the first base in an RLE k-mer, but
only 3 for the remaining k-1 bases since RLE k-mers do not contain
consecutive bases. Thus, 4×3k-1 is equal to the
total number of RLE k-mers.
With these default values, the total number of distinct
markers is approximately 0.1×4×39≈7900.
Options are also provided to read from a file the RLE k-mers to be used as markers. This permits experimentation with marker k-mers chosen in ways other than random. To date, no marker selection algorithm has proven more successful than random selection but it is very possible that assembly quality can be improved by non-random selection of markers.
The only constraint used in selecting k-mers to be used as markers is that if a k-mer is a marker, its reverse complement should also be a marker. This makes it easy to construct the marker representation of the reverse complement of a read from the marker representation of the original read. It also ensures strand symmetry in some of the computational steps.
Below is the run-length representation of a portion of a read and its markers, as displayed by the Shasta http server.

Marker alignments
The marker representation is a sequence
The marker representation of a read is a sequence in an alphabet consisting of the marker ids. This sequence is much shorter than the original sequence of the read but uses a much larger alphabet. For example, with default Shasta assembly parameters, the marker representation is 10 times shorter than the run-length encoded read sequence, or about 14 times shorter than the raw read sequence. Its alphabet has around 8000 symbols, many more than the 4 symbols that the original read sequence uses.Because the marker representation of a read is a sequence, we can compute an alignment of two reads directly in marker representation. Computing an alignment in this way has two important advantages:
- The shorter sequences and larger alphabet make the alignment much faster to compute.
- The alignment is insensitive to read errors in the portions that are not covered by any marker.
Alignment matrix
Consider two sequences on any alphabet, sequence x with nx symbols xi (i=0,...nx-1) and sequence y with ny symbols yj (j=0,...ny-1). The alignment matrix of the two sequences, , Aij, is a nx×ny matrix with elements
Aij = δxiyj
or, in words, Aij is 1 if xi=yj and 0 otherwise. (The Shasta assembler never explicitly constructs alignment matrices except for display when requested interactively. Alignment matrices are used here just for illustration).
In portions where the sequences x and y are perfectly aligned, the alignment matrix consists of a matrix diagonal set to 1. Most of the remaining elements will be 0, but many can be 1 just because the same symbol appears at two unrelated locations in the two sequences.
Alignment matrix in raw base representation
The picture below shows a portion of a typical alignment matrix of two reads in their representation as a raw base sequence (not the run-length encoded representation) and a computed optimal alignment. This is for illustration only, as the Shasta assembler never constructs such a matrix, except when requested interactively.
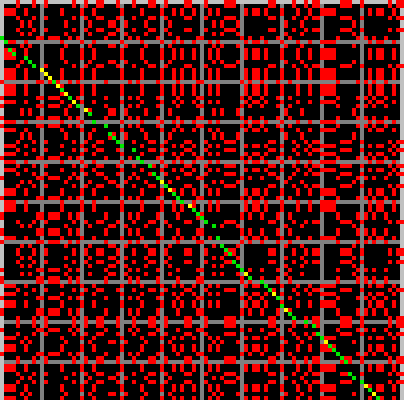
Here, elements of the alignment matrix are colored as follows:
- Red dots are alignment matrix elements that are 1 but that are not part of the computed optimal alignment.
- Green dots are alignment matrix elements that are 1 and that are part of the computed optimal alignment.
- Yellow matrix elements are alignment matrix elements that are 0 but that were computed to be part of the optimal alignment as mismatching alignment positions.
- Black or grey matrix elements are matrix elements that are 0 and that were not computed to be part of the optimal alignment. Grey is used instead of black every 10 bases to facilitate counting bases in the figure.
On average, about 25% of the matrix elements are 1, simply because the alphabet has 4 symbols. Because of the large fraction of 1 elements and because of the high error rate, it would be hard to visually locate the optimal alignment in this picture, if it was not highlighted using colors. This is emphasized by the following figure, which represents the same matrix, but with all 1 elements now colored red and all 0 elements black.

Note that the alignment matrix contains frequent square or rectangular blocks of 1 elements. They correspond to homopolymer run. A square block indicates that the two reads agree on the length of that homopolymer run, and a non-square rectangular block indicates that the two reads disagree. If we were using run-length encoding for this picture, these blocks would all collapse to a single matrix element.
Alignment matrix in marker representation
For comparison, the picture below shows a portion of the alignment matrix of two reads, in marker representation, as displayed by the Shasta http server. In this alignment matrix, the marker ordinal on the first read is on the horizontal axis (increasing towards the right) and the marker ordinal for the second read (increasing towards the bottom) is on the vertical axis. Here, matrix elements that are 1 are displayed in green or red. The ones in green are the ones that are part of the optimal alignment computed by the Shasta assembler - see below for more information. The grey lines are drawn 10 markers apart from each other and their only purpose is to facilitate reading the picture - the corresponding matrix elements are 0. Shasta marker alignments only include matching markers and gaps. Mismatching markers are never included in the alignment.

Because of the much larger alphabet, matrix elements that are 1 but are not part of the optimal alignment are infrequent. In addition, each alignment matrix element here corresponds on average to a 13×13 block in the alignment matrix in raw base sequence shown above. The portion of alignment matrix in marker space shown here covers about 120 markers or about 1500 bases in the original representation of the read, compared to only about 100 bases in the alignment matrix in raw representation shown above. For these reasons, the marker representation is orders of magnitude more efficient than the raw base representation when computing read alignments.
Alignment metrics
Shasta uses several metrics to characterize alignments in marker space. As shown in the picture above, a typical alignment consists of stretches of consecutive aligned markers intermixed with gaps in the alignment. Call xi (i=0,...,N-1) the aligned marker ordinals on the first oriented read and yj (j=0,...,N-1) the aligned marker ordinals on the second oriented read, where N is the number of aligned markers. That is, xi and yi are the coordinates of the green points in the alignment matrix as depicted above. Also, call Nx the number of markers in the first oriented read and Ny the number of markers in the second oriented read. Nx and Ny are the horizontal and vertical sizes, respectively, of the alignment matrix.
With this notation, we define the following metrics for a marker alignment. For each metric, there is a command line option that controls the minimum or maximum value of the metric for an alignment to be considered valid and used in the assembly. These options can be used to control the quality of marker alignments to be used during assembly.
- N
is the number of aligned markers.
An alignment is only used if N
is at least equal to the value of
--Align.minAlignedMarkerCount. This option can be used to discard alignments that are too short to be considered reliable. -
For each oriented read in the alignment, the
alignedFractionis defined as the ratio of the number of aligned markers over the length of the range of ordinals covered by the alignment, that is N / (xN-1 - x0 + 1) for the first oriented read and N / (yN-1 - y0 + 1) for the second oriented read. The lesser of thealignedFractionof the two oriented reads is called thealignedFractionof the alignment. It is a measure of the accuracy of the alignment. An alignment is only used ifalignedFraction is at least equal to the value of --Align.minAlignedFraction.- The maximum number of markers skipped by any alignment gap (on either ot the two oriented reads) is called
skipand is defined as the maximum value of max(xi+1 - xi, yi+1 - yi) computed over i=1,...,N-1. Note that, with this definition, gaps at the beginning and end of the alignment are not considered (but seetrimbelow). An alignment is only used ifskipis at most equal to the value of--Align.maxSkip. This option can be used to discard alignments containing long gaps.- Even when an alignment gap occurs, successive aligned markers tend to be approximately on the same diagonal of the alignment matrix. This reflects the fact that, even when sequencing errors occur, the number of bases read is likely to still be approximately correct. The maximum diagonal shift between successive aligned markers is called
driftand is defined as the maximum value of the absolute value of (xi+1 - yi+1) - (xi - yi) computed over i=1,...,N-1. Note that, in this case too, alignment gaps at the beginning and end of the alignment are not considered. An alignment is only used ifdriftis at most equal to the value of--Align.maxDrift. This option can be used to discard alignments containing gaps with large diagonal shift.- An alignment should always begin near the beginning of at least one of the two oriented reads, but we need to allow for an initial incorrect portion at the beginning of each read. The minimum over the two oriented reads of the number of markers skipped at the beginning of the alignment, min(x0, y0) is called the
leftTrim. Similarly, the minimum over the two oriented reads of the number of markers skipped at the end of the alignment, min(Nx - 1 - xN-1, Ny - 1 - yN-1) is called therightTrim. The lesser ofleftTrimandrightTrimis calledtrimAn alignment is only used iftrim. is at most equal to the value of--Align.maxTrim. - The maximum number of markers skipped by any alignment gap (on either ot the two oriented reads) is called
Computing optimal alignments in marker representation
To compute the optimal alignment highlighted in green in the above figure,
one of the following methods is used,
under control of command line option
--Align.alignMethod:
--Align.alignMethod 0selects the legacy Shasta algorithm. Do not use for production assemblies.--Align.alignMethod 1computes marker alignments using SeqAn. Do not use for production assemblies.--Align.alignMethod 3(the default choice) uses a faster two-step algorithm that also uses SeqAn and achieves better performance using a combination of downsampling and banded alignments.
The computation of marker alignments for all alignment candidates
found by the
LowHash algorithm
is one of the most expensive
phases of an assembly, and typically takes around half of
total elapsed time.
For this reason, it is recommended that only
the default --Align.alignMethod 3
be used for production assemblies.
--Align.alignMethod 0
This selects
a simple alignment algorithm
on the marker representations of the two reads to be aligned.
Do not use this method for production assemblies,
as the default alignment method selected by
--Align.alignMethod 3
provides better performance and accuracy.
This algorithm effectively constructs an optimal path in the alignment matrix,
but uses some heuristics to speed up the computation:
- The maximum number of markers that an alignment
can skip on either read is limited to a maximum,
under control of assembly parameter
--Align.maxSkip(default value 30 markers, corresponding to around 400 bases when all other Shasta parameters are at their default). This reflects the fact that Oxford Nanopore reads can often have long stretches in error. In the alignment matrix shown above, there is a skip of about 20 markers (2 light grey squares) following the first 10 aligned markers (green dots) on the top left. - The maximum number of markers that an alignment
can skip at the beginning or end of a read is limited to a maximum,
under control of assembly parameter
--Align.maxTrim(default value 30 markers, corresponding to around 400 bases when all other Shasta parameters are at their default). This reflects the fact that Oxford Nanopore reads often have an initial or final portion that is not usable. - To avoid alignment artifacts,
marker k-mers that are too frequent in either of the two reads
being aligned are not used in the alignment computation.
For this purpose, the Shasta assembler uses a criterion based
on an absolute number of occurrences of marker k-mers in the two reads,
although a relative criterion (occurrences per Kb) may be more appropriate.
The current absolute frequency threshold is under control of assembly parameter
--Align.maxMarkerFrequency(default 10 occurrences).
--Align.alignMethod 1
This causes marker alignments to be computed using
SeqAn overlap alignments.
This guarantees an optimal alignment, but is computationally very expensive,
with cost proportional to the size of the alignment matrix,
therefore O(N2) in the number of markers
in the two reads being aligned.
Do not use this method for production assemblies,
as the default alignment method selected by
--Align.alignMethod 3 provides much better performance
with minimal degradation in accuracy.
--Align.alignMethod 3
This is the default alignment method used by Shasta and provides a good combination of performance and accuracy. It uses the SeqAn in a two-step process:
- In a first step, SeqAn is used to compute an overlap alignment between two downsampled versions of the marker sequences of the reads to be aligned. This is O(N2) in the number of markers in the two reads being aligned, but still reasonably fast because of downsampling.
- In a second step, SeqAn is used to compute a banded alignment of the marker representation of the two reads. The position and width of the band is obtained from the downsampled alignment computed in the first step.
Finding overlapping reads
Even though computing read alignments in marker representation is fast, it still is not feasible to compute alignments among all possible pairs of reads. For a human-size genome with ≈106-107 reads, the number of pairs to consider would be ≈1012-1014, and even at 10-3 seconds per alignment the compute time would be ≈109-1011 seconds, or ≈107-109 seconds elapsed time (≈102-104 days) when using 128 virtual processors.
Therefore some means of narrowing down substantially the number of pairs to be considered is essential. The Shasta assembler uses for this purpose a slightly modified MinHash scheme based on the marker representation of reads.
For a general description of the MinHash algorithm see the Wikipedia article or this excellent book chapter. In summary, the MinHash algorithm takes as input a set of items each characterized by a set of features. Its goal is to find pairs of the input items that have a high Jaccard similarity index - that is, pairs of items that have many features in common. The algorithm proceeds by iterations. At each iteration, a new hash table is created and a hash function that operates on the feature set is selected. For each item, the hash function of each of its features is evaluated, and the minimum hash function value found is used to select the hash table bucket that each item is stored in. It can be proven that the probability of two items ending up in the same bucket equals the Jaccard similarity index of the two items - that is, items in the same bucket are more likely to be highly similar than items in different buckets. The algorithm then adds to the pairs of potentially similar items all pairs of items that are in the same bucket.
When all iterations are complete, the probability that a pair of items was found at least once is an increasing function of the Jaccard similarity of the two items. In other words, the pairs found are enriched for pairs that have high similarity. One can now consider all the pairs found (hopefully a much smaller set than all possible pairs) and compute the Jaccard similarity index for each, then keep only the pairs for which the index is sufficiently high. The algorithm does not guarantee that all pairs with high similarity will be found - only that the probability of finding all pairs is an increasing function of their similarities.
The algorithm is used by Shasta with items being oriented reads (a read in either original or reverse complemented orientation) and features being consecutive occurrences of m markers in the marker representation of the oriented read. For example, consider an oriented read with the following marker representation:
18,45,71,3,15,6,21If m is selected equal to 4 (the Shasta default, controlled by assembly parameter
--MinHash.m),
the oriented read is assigned the following features:
(18,45,71,3) (45,71,3,15) (71,3,15,6) (3,15,6,21)
From the picture above of an alignment matrix in marker representation, we see that streaks of 4 or more common consecutive markers are relatively common. We have to keep in mind that, with Shasta default parameters, 4 consecutive markers span an average 40 bases in run-length encoding or about 52 bases in the original raw base representation. At a typical error rate of around 10%, such a portion of a read would contain on average 5 errors. Yet, the marker representation in run-length space is sufficiently robust that these common "features" are relatively common despite the high error rate. This indicates that we can expect the MinHash algorithm to be effective in finding pairs of overlapping reads.
However, the MinHash algorithm has a feature that is undesirable for our purposes: namely, that the algorithm is good at finding read pairs with high Jaccard similarity index. For two sets X and Y, the Jaccard similarity index is defined as the ratio
J = |X∩Y| / |X∪Y|Because the read length distribution of Oxford Nanopore reads is wide, it is common to have pairs of reads with very different lengths. Consider now two reads with lengths nx and ny, with nx<ny, that overlap exactly over the entire length nx. The Jaccard similarity is in this case given by nx/ny < 1. This means that, if one of the reads in a pair is much shorter than the other one, their Jaccard similarity will be low even in the best case of exact overlap. As a result, the unmodified MinHash algorithm will not do a good job at finding overlapping pairs of reads with very different lengths.
For this reason, the Shasta assembler uses a small modification to the MinHash algorithm: instead of just using the minimum hash for each oriented read for each iteration, it keeps all hashes below a given threshold. Each oriented read can be stored in multiple buckets, one for each low hash encountered. This has the effect of eliminating the bias against pairs in which one read is much shorter than the other. The modified algorithm is referred to as LowHash in the Shasta source code. It is effectively equivalent to an indexing approach in which we index all features with low hash.
The LowHash algorithm is controlled by the following assembly parameters:
-
--MinHash.m(default 4): the number of consecutive markers that define a feature. -
--MinHash.hashFraction(default 0.01): The fraction of hash values that count as "low". -
--MinHash.minHashIterationCount(default 10): The number of iterations. -
--MinHash.maxBucketSize(default 10): The maximum number of items for a bucket to be considered. Buckets with more than this number of items are ignored. The goal of this parameter is to mitigate the effect of common repeats, which can result in buckets containing large numbers of unrelated oriented reads. -
--MinHash.minBucketSize(default 0): The minimum number of items for a bucket to be considered. Buckets with less than this number of items are ignored because MinHash features in these buckets are likely to be in error. -
--MinHash.minFrequency(default 2): the number of times a pair of oriented reads has to be found to be considered and stored as a possible pair of overlapping reads.
Initial assembly steps
Initial steps of a Shasta assembly proceed as follows.
If the assembly is setup for
best performance
(--memoryMode filesystem --memoryBacking 2M
if using the Shasta executable), all data structures
are stored in memory, and no disk activity takes
place except for initial loading of the input reads,
storing of assembly results, and storing a small number
of small files with useful summary information.
- Input reads are read from Fasta files and converted
to run-length representation. Read shorter
than the read length cutoff
--Reads.minReadLengthbases (default 10000) are discarded. Reads that contain bases with repeat counts greater than 255 are also discarded. This is a consequence of the fact that repeat counts are stored using one byte, and therefore there would be no way to store such reads. Reads with such long repeat counts are extremely rare, however, and when they occur they are of suspicious quality. - In addition, if a non-zero value is specified for
--Reads.desiredCoverage, the read length cutoff is further increased until coverage is just above the specified number of bases. - K-mers to be used as markers are randomly selected.
- Occurrences of those marker k-mers in all oriented reads are found.
- Reads are aligned to their reverse complement to determine if they are palindromic. Palindromes are flagged and ignored during assembly.
- The LowHash algorithm finds candidate pairs of overlapping oriented reads.
- A marker alignment is computed for each candidate pair of oriented reads. If the alignment metrics are sufficiently good, the alignment is stored for use in the assembly.
Read graph
Using the methods covered so far, an assembly has created a list of pairs of oriented reads, each pair having a plausible marker alignment. How to use this type of information for assembly is a classical problem with a standard solution (Myers, 2005), the string graph. However, the prescriptions in the Myers paper cannot be directly used here, mostly due to the marker representation.
Shasta proceeds by creating a Read Graph, an undirected
graph in which each vertex corresponds to an oriented read.
A subset of the available alignments is selected using one of two
methods described below, under control of command line option
--ReadGraph.creationMethod.
For each selected alignment, an undirected edge is created in the read
graph between the vertices corresponding to the oriented reads
in the selected alignment.
If we used all of the available alignments to create read graph edges, the resulting read graph would suffers from high connectivity in repeat regions, due to spurious alignments between oriented reads originating in different but similar regions of the genome. Therefore it is important to select alignments to be used in a way that minimizes these spurious alignments while keeping true ones as much as possible.
Note that each read contributes two vertices to the read graph, one in its original orientation, and one in reverse complemented orientation. Therefore the read graph contains two strands, each strand at full coverage. This makes it easy to investigate and potentially detect erroneous strand jumps that would be much less obvious if using alternative approaches with one vertex per read.
An example of a portion of the read graph, as displayed
by the Shasta http server, is shown here.

Even though the graph is undirected, edges that correspond to overlap alignments are drawn with an arrow that points from the leftmost oriented read to the rightmost one. Edges that correspond to containment alignments are drawn in red and without an arrow. Vertices are drawn with area proportional to the length of the corresponding reads.
The linear structure of the read graph successfully reflects the linear arrangement of the input reads and their origin on the genome being assembled.
However, deviations from the linear structure can
occur in the presence of long repeats,
typically for high similarity segment duplications:
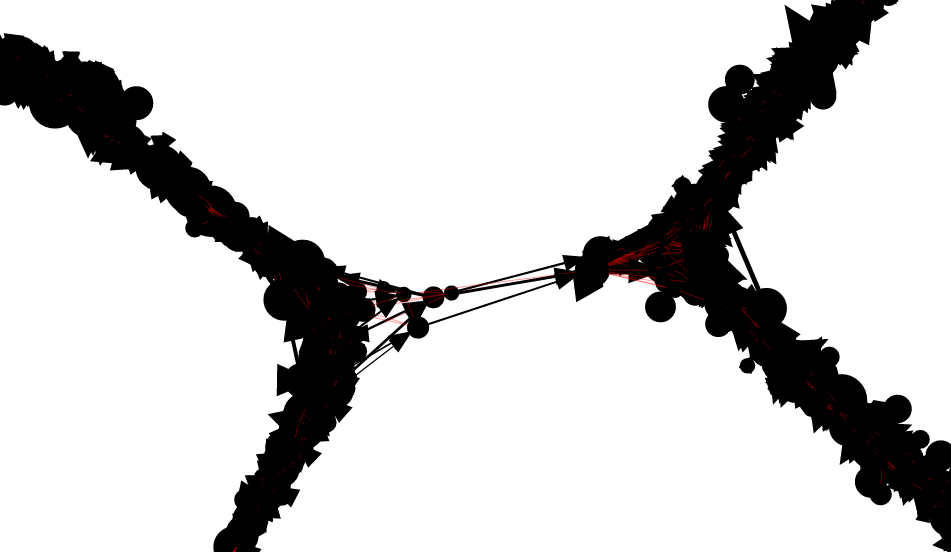
Obviously incorrect connections can destroy the linear structure of assembled sequence. This is dealt with later in the assembly process.
Read graph creation method 0
When --ReadGraph.creationMethod 0
(the default) is selected, the Shasta assembler uses a very simple method to select
alignments: it only keeps a
k-Nearest-Neighbor
subset of the alignments.
That is, for each vertex (oriented read)
it only keeps the best k alignments,
as measured by the number of aligned markers.
The number of edges k kept
for each vertex is controlled by assembly option
--ReadGraph.maxAlignmentCount,
with a default value of 6.
Note that, despite the k-Nearest-Neighbor subset,
it remains possible for a vertex to have a degree
more than k.
Read graph creation method 2
When --ReadGraph.creationMethod 2
is selected, a more sophisticated approach,
developed by Ryan Lorig-Roach at U. C. Santa Cruz,
is used.
Shasta first inspects all stored alignments to compute
statistical distributions (histograms) for the five alignment
metrics in the table below.
Alignments for which one of these metrics is worse than a set percentile
are discarded. The threshold percentiles are controlled by
the command line options shown in the table.
| Alignment metric | Percentile option | Default value |
|---|---|---|
| Number of aligned markers | --ReadGraph.markerCountPercentile
| 0.015 |
| Aligned marker fraction | --ReadGraph.alignedFractionPercentile
| 0.12 |
| Maximum number of markers skipped | --ReadGraph.maxSkipPercentile
| 0.12 |
| Maximum diagonal drift | --ReadGraph.maxDriftPercentile
| 0.12 |
| Number of markers trimmed | --ReadGraph.maxTrimPercentile
| 0.015 |
The process than continues with the same
k-Nearest-Neighbor procedure used with
--ReadGraph.creationMethod 0,
but only considering alignments that were not ruled
out by above percentile criteria.
Because of its adaptiveness to alignment characteristics,
this method is more robust and less sensitive
to the choice of alignment criteria
and results in more accurate and more contiguous assemblies.
However, when using --ReadGraph.creationMethod 2
it is important to use very liberal choices for the
options that control alignment quality because
bad alignments will be discarded anyway using the
percentile criteria.
An example of assembly options with --ReadGraph.creationMethod 2
is provided in Shasta configuration files
Nanopore-Sep2020.conf and
Nanopore-UL.Sep2020.conf.
These files apply to Oxford Nanopore reads created
with base caller Guppy version 3.6.0 or newer
(standard and ultra-long version, respectively).
They are available in the
conf directory in the source code tree
or in a Shasta build.
Marker graph
Consider a read whose marker representation is:
a b c d eWe can represent this read as a directed graph that describes the sequence in which its markers appear:
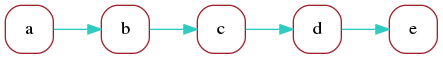
This is not very useful but illustrates the simplest form of a marker graph as used in the Shasta assembler. The marker graph is a directed graph in which each vertex represents a marker and each edge represents the transition between consecutive markers. We can associate sequence with each vertex and edge of the marker graph:
- Each vertex is associated with the sequence of the corresponding marker.
- If the markers of the source and target vertex of an edge do not overlap, the edge is associated with the sequence intervening between the two markers.
- If the markers of the source and target vertex of an edge do overlap, the edge is associated with the overlapping portion of the marker sequences.
Consider now a second read with the following marker
representation, which differs from the previous one
just by replacing marker c with x:
a b x d e
The marker graph for the two reads is:
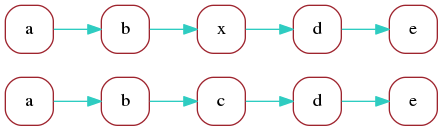
In the optimal alignment of the two reads, markers
a, b, d, e are aligned. We can redraw the marker graph
grouping together vertices that correspond to aligned markers:
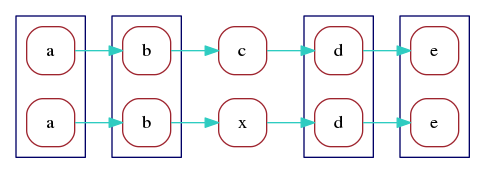
Finally, we can merge aligned vertices to obtain a marker graph describing the two aligned reads:

Here, by construction, each vertex still has a unique
sequence associated with it - the common sequence
of the markers that were merged
(however the corresponding repeat counts
can be different for each contributing read).
An edge, on the other hand, can have different sequences
associated with it, one corresponding to each
of the contributing reads.
In this example, edges a->b
and d->e have two contributing reads,
which can each have distinct sequence between
the two markers.
We call coverage of a vertex or edge the number of reads "contributing"
to it. In this example, vertices a, b, d, e have coverage 2,
and vertices c, x have coverage 1.
Edges a->b
and d->e have coverage 2, and the remaining edges have coverage 1.
The construction of the marker graph was illustrated above for two reads, but the Shasta assembler constructs a global marker graph which takes into account all oriented reads:
- The process starts with a distinct vertex for each marker of each oriented read. Note that at this stage the marker graph is large (≈ 2×1010 vertices for a human assembly using default assembly parameters).
- For each marker alignment corresponding to an edge of the read graph, we merge vertices corresponding to aligned markers.
- Of the resulting merged vertices, we remove those whose
coverage is too low or too high, indicating that the contributing
reads or some of the alignments involved are probably in error.
This is controlled by assembly parameters
MarkerGraph.minCoverage(default 10) andMarkerGraph.maxCoverage(default 100), which specify the minimum and maximum coverage for a vertex to be kept. - Edges are created. An edge
v0->v1is created if there is at least a read contributing to bothv0andv1and for which all markers intervening betweenv0andv1belong to vertices that were removed.
Given the large number of initial vertices involved, this computation is not trivial. To allow efficient computation in parallel on many threads, a lock-free implementation of the disjoint data set data structure, as first described by Anderson and Woll (1991), Anderson and Woll (1994), is used for merging vertices. Some code changes were necessary to permit large numbers of vertices, as the initial implementation by Wenzel Jakob only allowed for 32-bit vertex ids.
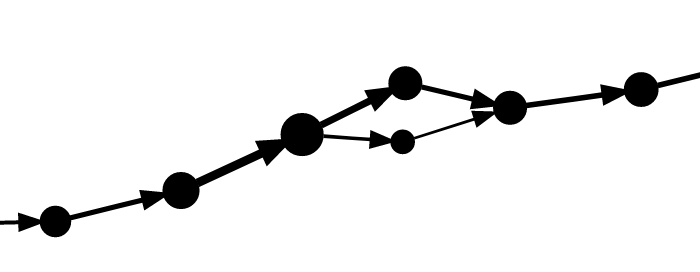
A portion of the marker graph, as displayed by the Shasta http server, is shown here:

Each vertex, shown in green, contains a list of the oriented reads that contribute to it. Each oriented read is labeled with the read id (a number) followed by "-0" for original orientation and "-1" for reverse complemented orientation. Each vertex is also labeled with the marker sequence (run-length encoded). Edges are drawn with an arrow whose thickness is proportional to edge coverage and labeled with the sequence contributed by each read. In edge labels, a sequence consisting of a number indicates the number of overlapping bases between adjacent markers.
On a larger scale and with less detail, a typical portion of the marker graph looks like this:

Here, each vertex is drawn with a size proportional to its coverage. Edge arrows are again displayed with thickness proportional to edge coverage. The marker graph has a linear structure with a dominant path and side branches due to errors.
Assembly graph
The Shasta assembly process also uses a compact representation of the marker graph, called the assembly graph, in which each linear sequence of edges is replaced by a single edge. For example, this marker graph

can be represented as an assembly graph as follows. Colors were chosen to indicate the correspondence to marker graph edges:

The length of an edge of the assembly graph is defined as the number of marker graph edges that it corresponds to. For each edge of the assembly graph, average coverage is also computed, by averaging the coverage of the marker graph edges it corresponds to.
Using the marker graph to assemble sequence
The marker graph is a partial description of the multiple sequence alignment between reads and can be used to assemble a consensus sequence. One simple way to do that is to only keep the "dominant" path in the graph, and then move on that path from vertex to edge to vertex, assembling run-length encoded sequence as follows:
- On a vertex, all reads have the same sequence, by construction: the marker sequence associated with the vertex. There is trivial consensus among all the reads contributing to a vertex, and the marker sequence can be used directly as the contribution of the vertex to assembled sequence.
- For edges, there are two possible situations plus a hybrid case:
- 2.1. If the adjacent markers overlap, in most cases, all contributing reads have the same number of overlapping bases between the two markers, and we are again in a situation of trivial consensus, where all reads contribute the same sequence, which also agrees with the sequence of adjacent vertices. In cases where not all reads are in agreement on the number of overlapping bases, only reads with the most frequent number of overlapping bases are taken into account.
- 2.2. If the adjacent markers don't overlap, then each read can have a different sequence between the two markers. In this situation, we compute a multiple sequence alignment of the sequences and a consensus using the Spoa library. The multiple sequence alignment is computed constrained at both ends, because all reads contributing to the edge have, by construction, identical markers at both sides.
- 2.3. A hybrid situation occasionally arises, in which some reads have the two markers overlapping, and some do not. In this case, we count reads of the two kinds and discard the reads of the minority kind, then revert to one of the two cases 2.1 or 2.2 above.
This is the process used for sequence assembly by the current Shasta implementation. It requires a process to select and define dominant paths, which is described in the next section. It is algorithmically simple, but its main shortcoming is that it does not use for assembly reads that contribute to the abundant side branches. This means that coverage is lost, and therefore the sequence of assembled accuracy is not as good as it could be if all available coverage was used. Means to eliminate this shortcoming and use information from the side branches of the marker graph could be a subject of future work on the Shasta assembler.
Assembling repeat counts
The process described above works with run-length encoded sequence and therefore assembles run-length encoded sequence. The final step to create raw assembled sequence is to compute the most likely repeat count for each sequence position in run-length encoding. In Shasta 0.1.0 this was done by choosing as the most likely repeat count the one that appears the most frequently in the reads that contributed to each assembled position.
The latest version of the Shasta assembler supports three different options
for that, under control of command line option
--Assembly.consensusCaller:
--Assembly.consensusCaller Modal(the default) requests the same algorithm used in Shasta 0.1.0, that is, the most frequent repeat count seen at each base position is used in the assembly.--Assembly.consensusCaller Medianrequests an algorithm that stores in the assembly the median repeat count seen at each base position.--Assembly.consensusCaller Bayesian:namerequests a Bayesian algorithm to determine the most likely repeat count.namecan be one of the following:- One of the following built-in Bayesian models:
guppy-2.3.1-ato use a Bayesian model optimized for the Guppy 2.3.1 base caller.guppy-3.0.5-ato use a Bayesian model optimized for the Guppy 3.0.5 base caller.guppy-3.4.4-ato use a Bayesian model optimized for the Guppy 3.4.4 base caller.guppy-3.6.0-ato use a Bayesian model optimized for the Guppy 3.6.5 base caller.r10-guppy-3.4.8-ato use a Bayesian model optimized for r10 reads and the Guppy 3.4.8 base caller.bonito-0.3.1-ato use a Bayesian model optimized for the Bonito 0.3.1 base caller.guppy-5.0.7-bto use a Bayesian model optimized for the Guppy 5.0.7 base caller. There is also an older and less accurate modelguppy-5.0.7-a.
- The name of a configuration file
describing the Bayesian model to be used,
which can be specified as a relative or absolute path.
Earlier versions of Shasta required an absolute path, but this
is no longer the case.
Sample configuration files are available in
shasta/conforshasta-install/conf. They are namedSimpleBayesianConsensusCaller-*.csv.
- One of the following built-in Bayesian models:
--Assembly.Modal.
Some testing showed a significant decrease of false positive indels
in assembled sequence compared to Shasta 0.1.0 behavior, corresponding to
--Assembly.consensusCaller Modal. This testing was done
for reads called using Guppy 2.3.5. However, even using
--Assembly.consensusCaller Bayesian:guppy-2.3.1-a
resulted in significant improvement over
--Assembly.consensusCaller Modal, indicating
that the Bayesian model is somewhat resilient to
discrepancies between the reads used to construct the Bayesian model
and the reads being assembled.
Testing also showed that
--Assembly.consensusCaller Median is generally inferior to
--Assembly.consensusCaller Modal.
Software to create a new Bayesian model for a new data type is available at
https://github.com/rlorigro/runlength_analysis_cpp.
This creates a configuration file containing the definition of
the newly created Bayesian model, in a csv format that can be used directly in Shasta
via
--Assembly.consensusCaller Bayesian:fileName.
Selecting assembly paths
The sequence assembly procedure described in the previous section
can be used to assemble sequence for any path in the marker graph.
This section describes the selection of paths for assembly
in the current Shasta implementation.
This is done by a series of steps that "remove" edges
(but not vertices) from the marker graph until the
marker graph consists mainly of linear sections
which can be used as the assembly paths.
For speed, edges are not actually removed but just marked
as removed using a set of flag bits allocated for this
purpose in each edge.
However, the description below will use the loose term remove
to indicate that an edge was flagged as removed.
This process consists of the following three steps, described in more detail in the following sections:
- Approximate transitive reduction of the marker graph.
- Pruning of short side branches (leaves).
- Removal of bubbles and super-bubbles.
Approximate transitive reduction of the marker graph
The goal of this step is to eliminate the side branches in the marker
graph, which are the result of errors.
Although
the number of side branches is substantially reduced thanks to the use of
run-length encoding, side branches are still abundant.
This step uses an approximate transitive reduction of the marker graph which only
considers reachability up to a maximum distance,
controlled by assembly parameter
MarkerGraph.maxDistance
(default 30 marker graph edges).
Using a maximum distance makes sure that the process remains computationally affordable,
and also has the advantage of not removing long-range edges in the marker graph,
which could be significant.
In detail, the process works as follows.
In this description, the edge being considered for removal
is the edge v0→v1 with source vertex v0
and target vertex v1.
The first two steps are not really part of the transitive reduction
but are performed by the same code for convenience.
- All edges with coverage less than or equal to
MarkerGraph.lowCoverageThresholdare unconditionally removed. The default value for this assembly parameter is 0, so this step does nothing when using default parameters. - All edges with coverage 1
and for which the only supporting read has a large marker skip
are unconditionally removed.
The marker skip of an edge, for a given read, is defined as the distance
(in markers) between the
v0marker for that read and thev1marker for the same read. Most marker skips are small, and a large skip is indicative of an artifact. Keeping those edges could result in assembly errors. The marker skip threshold is controlled by assembly parameterMarkerGraph.edgeMarkerSkipThreshold(default 100 markers). - Edges
with coverage greater than
MarkerGraph.lowCoverageThreshold(default 0) and less thanMarkerGraph.highCoverageThreshold(default 256), and that were not previously removed, are processed in order of increasing coverage. Note that with the default values of these parameters all edges are processed because edge coverage is stored using one byte and therefore can never be more than 255 (it is saturated at 255). For each edgev0→v1, a Breadth-First Search (BFS) in the marker graph is performed starting at source vertexv0and with a limit ofMarkerGraph.maxDistance(default 30) edges distance from vertexv0. The BFS is constrained to not use edgev0→v1. If the BFS reachesv1, indicating that an alternative path fromv0tov1exists, edgev0→v1is removed. Note that the BFS does not use edges that have already been removed, and so the process is guaranteed not to affect reachability. Processing edges in order of increasing coverage makes sure that low coverage edges are the most likely to be removed.
When the transitive reduction step is complete, the marker graph consists mostly of linear sections composed of vertices with in-degree and out-degree one, with occasional side branches and bubbles or superbubbles, which are handled in the next two phases described below.
Pruning of short side branches (leaves)
At this stage, a few iterations of pruning are done
by simply removing, at each iteration,
edge v0→v1 if
v0 has in-degree 0 (that is, is a backward-pointing leaf)
or v1 has out-degree 0 (that is, is a forward-pointing leaf).
The net effect is that all side branches of length
(number of edges)
at most equal to the number of iterations are removed.
This leaves the leaf vertex isolated, which causes no problems.
The number of iterations is controlled
by assembly parameter
MarkerGraph.pruneIterationCount
(default 6).
Removal of bubbles and superbubbles
The marker graph now consists of mostly linear section with occasional bubbles or superbubbles. Most of the bubbles and superbubbles are caused by errors, but some of those are due to heterozygous loci in the genome being assembled. Bubbles and superbubbles of the latter type could be used for separating haplotypes (phasing) - a possibility that will be addressed in future Shasta releases. However, the goal of the current Shasta implementation is to create a haploid assembly at all scales but the very long ones. Accordingly, bubbles and superbubbles at short scales are treated as errors, and the goal of the bubble/superbubble removal step is to keep the most significant path in each bubble or superbubble.
The figures below show typical examples of a bubble and superbubble in the marker graph.

The bubble/superbubble removal process is iterative.
Early iterations work on short scales, and late iterations
fork on longer scales.
Each iteration uses a length threshold
that controls the maximum number of marker graph edges for
features to be considered for removal.
The value of the threshold for each iteration
is specified using assembly parameter
MarkerGraph.simplifyMaxLength,
which consists of a comma-separated string of integer
numbers, each specifying the threshold for one iteration
in the process. The default value is
10,100,1000, which means that three iterations
of this process are performed.
The first iteration uses a threshold of 10 marker graph edges,
and the second and third iterations
use length thresholds of 100 and 1000 marker graph edges, respectively.
The last and largest of the threshold values used
determines the size of the smallest bubble or superbubble
that will survive the process.
The default 1000 markers is equivalent to roughly 13 Kb.
To suppress more bubble/superbubbles, increase
the threshold for the last iteration.
To see more bubbles/superbubbles, decrease
the length threshold for the last iteration, or remove the last iteration entirely.
The goal of the increased threshold values is to work
on small features at first, and on larger features in the later iterations.
The best choice of
MarkerGraph.simplifyMaxLength
is application dependent. The default value
is a reasonable compromise useful if one desires
a mostly haploid assembly with just some large heterozygous features.
Each iteration consists of two steps. The first removes bubbles and the second removes superbubbles. Only bubbles/superbubbles consisting of features shorter than the threshold for the current iteration are considered:
- Bubble removal
- An assembly graph corresponding to the current marker graph is created.
- Bubbles are located in which the length of all branches (number of marker graph edges) is no more than the length threshold at the current iteration. In the assembly graph, a bubble appears as a set of parallel edges (edges with the same source and target).
- In each bubble, only the assembly graph edge with the highest average coverage is kept. Marker graph edges corresponding to all other assembly graph edges in the bubble are flagged as removed.
- Superbubble removal:
- An assembly graph corresponding to the current marker graph is created.
- Connected components of the assembly graph are computed, but only considering edges below the current length threshold. This way, each connected component corresponds to a "cluster" of "short" assembly graph edges.
- For each cluster, entries into the cluster are located. These are vertices that have in-edges from a vertex outside the cluster. Similarly, exists are located (vertices that have out-edges outside the cluster).
- For each entry/exit pair, the shortest path is computed. However, in this case the "length" of an assembly graph edge is defined as the inverse of its average coverage - that is, the inverse of average coverage for all the contributing marker graph edges.
- Edges on each shortest path are marked as edges to be kept.
- All other edges internal to the cluster are removed.
When all iterations of bubble/superbubble removal are complete, the assembler creates a final version of the assembly graph. Each edge of the assembly graph corresponds to a path in the marker graph, for which sequence can be assembled using the method described above. Note, however, that the marker graph and the assembly graph have been constructed to contain both strands. Special care is taken during al transformation steps to make sure that the marker graph (and therefore the assembly graph) remain symmetric with respect to strand swaps. Therefore, the majority of assembly graph edges come in reverse complemented pairs, of which we assemble only one. It is however possible but rare for an assembly graph to be its own reverse complement.
Detangling
In many real-life situations, the assembly graph contains features like this one, called a tangle:

A tangle consists of an edge v0→v1 (depicted here in green) for which the following is true:
out-degree(v0) = 1
(No outgoing edges of v0 other than the green edge)
in-degree(v1) = 1
(No incoming edges of v1 other than the green edge)
in-degree(v0) > 1
(v0 has more than one incoming edge -
if that was not the case, the one incoming edge could be trivially merged with the green edge).
out-degree(v1) > 1
(v1 has more than one outgoing edge -
if that was not the case, the one outgoing edge could be trivially merged with the green edge).
In the most common case, in-degree(v0) = out-degree(v1) = 2, and this what the above picture and the following text assume, for clarity.
Because DNA structure is linear, this pattern expressed that two portions of sequence have similar portions (the green edge) which the assembly is not able to separate. However, in some cases, it is possible to separate or "detangle" these two copies of similar sequence.
If the green edge is short enough, there may be reads long enough to span it in its entirety. For those reads, we will be able to tell which of the edges on the left and right of the tangle they reach. Now, suppose we only see reads going from the blue edge on the left to the blue edge on the right, and reads going from the red edge on the left to the red edge on the right - but no reads crossing between blue and red edges. Then we can infer that one of the copies follows the blue edges on both sides, while the other copy follows the red edge. This allows us to detangle the two copies as follows:

Note that the sequence in the green edge was duplicated. This makes sense
because there are two copies of that sequence.
This detangling scheme was first proposed, in a different context,
by Pevzner et al. (2001).
It can be applied with no conceptual changes to the Shasta assembly graph.
Command line option
--Assembly.detangleMethod
is used to control detangling.
- --Assembly.detangleMethod 0 (default): detangling is turned off (not activated).
- --Assembly.detangleMethod 1: activates a strict form of detangling. Requires at least one read spanning the "red" edges on both sides, at least one read spanning the "blue" edges on both sides, and no reads crossing over between the red and blue edges.
- --Assembly.detangleMethod 2:
Activates a more relaxed form of detangling, controlled by three command line options
--Assembly.detangle.diagonalReadCountMin,
--Assembly.detangle.offDiagonalReadCountMax, and
--Assembly.detangle.offDiagonalRatio.
See the code (
shasta/src/AssemblyPathGraph2.cpp) for details of how these parameters control detangling.
Iterative assembly
Iterative assembly is a Shasta experimental feature
that provides the ability to separate haplotypes or similar copies of long repeats.
In preliminary tests on a human genome, it has been shown to provide some
amount of haplotype separation,
and therefore partially phased
diploid assembly, as well as improved ability to
resolve segmental duplications.
In current tests, it requires Ultra-Long (UL)
Nanopore reads created by base caller Guppy 3.6.0 or newer
at high coverage 80X.
The assembly options that were used in this process are captured in the configuration file
Nanopore-UL-iterative-Sep2020.conf.
The Shasta iterative assembly code is at an experimental stage
and therefore still subject to further
improvements and developments. In its implementation as of September 2020,
it operates as follows. In this description, copy refers
to a haplotype or a copy of a segmental duplication
or other long repeats.
- The assembly process runs as usual, without the final phases of bubble/superbubble removal and final sequence assembly.
-
Shasta then computes and stores the sequence of assembly graph edges
encountered by each oriented read, called the pseudo-path
of the oriented read.
These pseudo-paths contain detailed information on how each read
traverses each bubble/superbubble in the assembly graph.
Therefore, two oriented reads that originate from the same
copy(in the sense defined above) are likely to have largely concordant pseudo-paths. -
For each stored marker alignment between two oriented reads,
Shasta now computes an alignment of the pseudo-paths of the two reads.
If the alignment of the two pseudo-paths is not sufficiently good,
indicating that the two oriented reads originate from
distinct
copies, that marker alignment is flagged as not to be used. -
A new version of the read graph is created normally,
but excluding from consideration the marker alignments that
were flagged as not to be used in the previous step.
As a result,
in the new read graph edges are created preferentially
between oriented reads originating from the same
copy. Edges between oriented reads originating from distinctcopiesare less likely to be created. Therefore the resulting read graph has achieved some amount of separation between thecopies. -
The process is repeated a few times. At each iteration,
the new read graph achieves cleaner separation between
copies. - When the last iteration completes, the assembly process continues normally, with optional bubble/superbubble removal and detangling, followed by sequence assembly.
Phased diploid assembly
Note: a preliminary implementation of phased (mode 2) assembly was included in Shasta 0.8.0. For documentation of that preliminary implementation, get the tar file for Shasta 0.8.0. The documentation below describes the current, improved implementation of phased assembly.
Shasta provides a second assembly workflow specially tuned for phased diploid assembly. It still uses the same basic computational methods (MinHash/LowHash algorithm, read graph, marker graph), but adds a phasing process for diploid genomes that results in a good amount of haplotype separation in many cases.
Being specialized for the separation of haplotypes in a diploid assembly, this process is not effective at separating copies of segmental duplications, and for the same reason is not effective for genomes with higher ploidy. However, it can be used for genomes with mixed ploidy 1 and 2 such as the human genome. It will typically assemble two haplotypes at most locations in the genomes except segmental duplication, if:
- Coverage is high enough.
- The reads are long enough.
- Heterozygosity is not too low.
Because mode 2 assembly is less capable than mode 0 assembly (standard Shasta haploid assembly) in segmental duplications, mode 0 assembly can be more effective if assembly contiguity (N50) is the main goal, and separating haplotypes is not important.
Assembly mode 2 does not work by assigning haplotypes to reads. Rather, it assigns haplotypes to branches of heterozygous bubbles in the marker graph.
See the following sections for a description of the computational process used for mode 2 assembly and the output it creates.
Marker graph creation in mode 2 assembly
Mode 0 and mode 2 assembly generate marker graph vertices
in the same way: at the end of the disjoint sets process,
each disjoint set containing at least
--MarkerGraph.minCoverage
markers is allowed to generate a vertex if:
- It also contains at least
--MarkerGraph.minCoveragePerStrandmarkers on each strand. This is to avoid vertices with extreme strand bias, which are likely to correspond to systematic errors. - It does not contain more than one marker belonging to the same read and with the same orientation. This avoids some undesirable cyclic features in the marker graph.
A→B
between vertices A and B
is generated if there is at least one (oriented) read
that visits vertex B immediately
after vertex A, without visiting
any other vertices in between.
Then, edges are gradually removed by a process,
including transitive reduction and bubble/superbubble removal,
whose main goal is to keep contiguity unaltered.
This results in highly contiguous haploid assemblies (large N50).
In mode 2 assembly, the process of generating edges is in a way reversed:
rather than being very permissive in the initial creation of edges,
edges are initially created subject to strict coverage criteria
similar to those used for vertices,
under control of
--MarkerGraph.minEdgeCoverage
and
--MarkerGraph.minEdgeCoveragePerStrand.
This ensures that only edges with good read support are generated.
As an additional requirement, all oriented reads
that participate in an edge are required to have
exactly the same sequence on the edge
(or the same RLE sequence, if working with reads
in RLE representation).
If this is not the case, the edge is split.
This way, a bubble is generated (see below) if any two oriented reads have different sequences (or RLE sequences) in any vertex or edge.
This creates the following problem when working using reads in RLE representation. In that case, this process does not generate bubbles for heterozygous loci in which the two branches have the same RLE sequence. For example, consider a SNP A->G in which the two alleles are
AAAG AAGGFor both alleles, the RLE sequence is AG, so no bubble is generated, and that SNP could be missed. Nevertheless, with current ONT reads the benefits of RLE still outweigh this issue, and the current assembly configurations for phased assembly do use RLE.
However, the strict criteria for edge creation result in frequent breaks of contiguity at places where coverage is locally low due to errors. To avoid fragmented assemblies, this is addressed by adding after the facts a minimal number of secondary edges at the locations where breaks occur. For these secondary edges, the above coverage criteria are not used, but the logic to create secondary edges attempts to maximize coverage of the secondary edges that are created.
Because of the strict coverage criteria used, this results in a mostly linear marker graph. The transitive reduction used for mode 2 assembly is not used. And the bubble and superbubble removal process work differently in mode 2 assembly, as described below.
Heterozygous bubbles
The marker graph created as described above is mostly linear but has occasional bubbles caused by heterozygous loci. A bubble is a set of parallel edges in the marker graph. The number of parallel edges is the ploidy of the bubble.
Mode 2 assembly works under a diploidy assumption (ploidy is 2 everywhere). If any bubbles with ploidy greater than 2 are found, all branches except for the two strongest ones (most supporting reads) are kept.
The two branches of each diploid heterozygous bubble are ordered arbitrarily and labeled 0 and 1. We keep track of which reads appear in the marker graph edges of the two branches. In the next sections, the numbers of reads that appear on the two branches are called n0 and n1.
Diploid bubbles in the marker graph don't necessarily reflect the difference between haplotypes at a heterozygous locus and can be caused by errors. In fact, with current Nanopore reads most heterozygous bubbles are caused by errors. Therefore, heterozygous bubbles are handled in two steps in mode 2 assembly:
- In the first step, bubble removal, bubbles that are likely to be due to errors are removed by only keeping the strongest branch (that is, the one supported by the most reads).
- In the second step, phasing, a haplotype is assigned to each branch of each bubble that was not removed in the first step.
The first step is essential. Attempting to phase bubbles without first removing the ones caused by errors would render that phasing process noisy and unreliable.
In the phasing step, it is impossible to assign haplotypes globally. Rather, in the phasing step, heterozygous bubbles are partitioned into phasing components, and all bubbles in a phasing component are assigned a haplotype, which is valid only relative to other bubbles in the same component. Each phasing component corresponds to one "large bubble" in the phased representation of the assembly (see below). Occasionally, it can also correspond to more than one large bubble.
Superbubbles
Structures with connectivity more complex than simple bubbles (superbubbles) are generally present in the marker graph. Handling of these structures is currently limited to superbubbles with one entrance and one exit. The superbubble is replaced with a diploid bubble by choosing the two most likely paths in the superbubble.
Simple Bayesian model for a pair of diploid bubbles
The bubble removal and the phasing steps use a simple Bayesian model describing two diploid bubbles.
Given two diploid bubbles, bubble A and bubble B, we can use their read composition to tell how the haplotypes of the first bubble are related to the haplotypes of the second bubble. We can create a 2 by 2 phasing matrix that counts the number of common reads between each side of the two bubbles. That is, nij is the number of common reads between branch i of bubble A and branch j of bubble B. In an ideal error-free scenario, one of two following situations would occur:
- nij is diagonal, that is, n01 = n10 = 0. All common reads that visit branch 0 of bubble A also visit branch 0 of bubble B, and all common reads that visit branch 1 of bubble A also visit branch 1 of bubble B. In this case, we can say that the two bubbles are in-phase, that is, branch 0 of bubble A is on the same haplotype as branch 0 of bubble B, and branch 1 of bubble A is on the same haplotype as branch 1 of bubble B,
- nij has a zero diagonal, that is, n00 = n11 = 0. All common reads that visit branch 0 of bubble A also visit branch 1 of bubble B, and all common reads that visit branch 1 of bubble A also visit branch 0 of bubble B. In this case, we can say that the two bubbles are out-of-phase, that is, branch 0 of bubble A is on the same haplotype as branch 1 of bubble B, and branch 1 of bubble A is on the same haplotype as branch 0 of bubble B.
However, due to errors that can be deviations from this ideal behavior. In addition, if even one of the two bubbles is caused by errors the distribution of the reads between the branches of the two bubbles can be entirely random. To describe this quantitatively, we use a simple Bayesian model for the pair of diploid bubbles.
Here are some additional quantities we need below.
- The number of reads on branch 0 of bubble A that also appear on either branch of bubble B on is nA0 = n00 + n01.
- The number of reads on branch 1 of bubble A that also appear on either branch of bubble B on is nA1 = n10 + n11.
- The number of reads on branch 0 of bubble B that also appear on either branch of bubble A on is nB0 = n00 + n10.
- The number of reads on branch 1 of bubble B that also appear on either branch of bubble B on is nB1 = n01 + n11.
- The number of reads on the diagonal of nij is defined as ndiagonal = n00 + n11. This is the number of reads that suggest that the two bubbles are in-phase.
- The number of reads off the diagonal of nij is defined as noff-diagonal = n01 + n10. This is the number of reads that suggest that the two bubbles are out of phase.
- The number of reads that suggest the "best" of the in-phase and out of phase hypotheses (based on simple read counts) is defined as nconcordant = max(ndiagonal, noff-diagonal).
- The number of reads that suggest the "worst" of the in-phase and out of phase hypotheses (based on simple read counts) is defined as ndiscordant = min(ndiagonal, noff-diagonal).
- The total number of reads that appear on both bubbles is n = nA0 + nA1 = nB0 + nB1 = n00 + n01 + n10 + n11 = ndiagonal + noff-diagonal = nconcordant + ndiscordant
With the above values given and considered fixed, we now consider three possible hypotheses for the two bubbles:
- Random hypothesis: one or both of the two bubbles are caused by errors, and therefore reads visits the two bubbles in an entirely uncorrelated fashion.
- Ideal in-phase hypothesis: the two bubbles are in-phase, that is, branches 0 of the two bubbles are on the same haplotype, and similarly for branches 1. Under this ideal version of the in-phase hypothesis we also rule out all errors, and as a result, the phasing matrix is exactly diagonal.
- Ideal out-of-phase hypothesis: the two bubbles are out-of-phase, that is, branch 0 of bubble A is on the same haplotype as branch 1 of bubble B, and similarly for the other two haplotypes. Under this ideal version of the out-of-phase hypothesis we also rule out all errors, and as a result, the diagonal of the phasing matrix is exactly zero.
Random hypothesis
Under the random hypothesis, reads visit the two branches of the two bubbles in an entirely uncorrelated fashion. The probability that one of the n reads visits branch i of bubble A and branch j of bubble B is:
P(ij | random) = (nAi / n) (nBj / n) = nAi nBj / n 2
It can be easily verified that the sum of all four values for P(ij|random) equals 1, as it should.
Ideal in-phase hypothesis
Under the ideal in-phase hypothesis, reads visit the two bubbles just like in the random hypothesis, except that the off-diagonal elements are zero. Therefore P(ij | in-phase) can be obtained from P(ij | random) by setting the off-diagonal entries to zero and renormalizing the diagonal entries so they add up to 1. The result is:
P(ij | ideal in-phase) = δij nAi nBj / (nA0nB0 + nA1nB1)
Ideal out-of-phase hypothesis
The corresponding expression for the ideal out-of-phase hypothesis can be obtained in a similar way. The result is:
P(ij | ideal out-of-phase) = (1-δij) nAi nBj / (nA0nB1 + nA1nB0)
Non-ideal hypotheses
In reality, we have to consider non-ideal in-phase and out-of-phase hypotheses in which reads have probability ε>0 of visiting branches in the two bubbles that is inconsistent with the corresponding ideal hypothesis (a simple but perhaps oversimplistic assumption). Under these non-ideal hypotheses, the probability that one of the n reads visits branch i of bubble A and branch j of bubble B is
P(ij | in-phase) = (1 - ε) P(ij | ideal in-phase) + ε P(ij | ideal out-of-phase) =
P(ij | out-of-phase) = (1 - ε) P(ij | ideal out-of-phase) + ε P(ij | ideal in-phase) =
Bayesian model
With the above expressions available, we are in a position to construct a simple Bayesian model in which we evaluate posterior probability ratios of the above hypotheses conditional to the observed distribution of reads visiting the two branches of each bubble.
We use "neutral" assumptions on prior probabilities:
Pprior(in-phase) = Pprior(out-of-phase) = Pprior(random)
Posterior probability ratios are then given by:
log[Pposterior(in-phase) / Pposterior(random)] = ∑ij nij log[P(ij | in-phase) / P(ij | random)]
Here, the sums over i and j run over values 0 and 1, that is, over the entire 2 by 2 phasing matrix. In the expressions on the right, nij are entries of the phasing matrix, and the remaining expressions have been evaluated in the above sections. Because we assumed ε>0, all terms in the above expressions are non-singular.
Extension of the Bayesian model to two sets of diploid bubbles
Consider two disjoint sets of diploid bubbles, S0 and S1. Assume the bubbles in each of the two sets have already been phased relative to each other, that is, we know how the haplotypes of each bubble in each of the set correspond to haplotypes of other bubbles in the same set. The Bayesian model described in the previous section can be extended to apply to the phasing of S0 relative to S1. This can be useful, as it is possible that there is insufficient evidence to phase individual bubbles of S0 relative to bubbles of S1, while sufficient combined evidence still is present to support phasing the two sets relative to each other.
Such an extended Bayesian model permits a hierarchical approach to phasing, described in more details below, in which sets of bubbles are iteratively phased and combined into larger phased sets.
The phasing matrix for the extended Bayesian model can be constructed similarly for the case of a single pair of bubbles. The phasing matrix counts distinct reads, so if a read appears in multiple bubbles of the two sets, it is counted only once.
Phasing graph
Phasing information for a set of diploid bubbles can be described using a phasing graph, an undirected graph in which each vertex represents a bubble or a set of bubbles already phased relative to each other. The phasing graph is used in two steps, described in more detail below.
- The goal of the first step, bubble removal, is to remove bubbles that are likely to be the result of errors. In this step, each vertex of the phasing graph corresponds to a single bubble rather than a set of bubbles.
- The goal of the second step, phasing, is to assign a haplotype to each branch of each bubble. This is done hierarchically by iteratively phasing groups of bubbles relative to each other - see below for more details.
The phasing graph is used in both steps, with some differences.
An edge between two vertices can be created if the two corresponding bubbles or sets of bubbles have enough common reads to permit phasing. The precise criteria for edge creation are different for bubble removal and phasing, but in both cases each edge stores the nij phasing matrix for the bubbles corresponding to the vertices it joins. But in both cases, we enforce a minimum allowed value for nconcordant and a maximum allowed value for ndiscordant. This reflects the fact that, for an edge to carry sufficient information, we would like nconcordant to be high and ndiscordant to be small. These thresholds are controlled by the following command line options:
- For bubble removal,
--Assembly.mode2.bubbleRemoval.minConcordantReadCountand--Assembly.mode2.bubbleRemoval.maxDiscordantReadCount. - For phasing,
--Assembly.mode2.phasing.minConcordantReadCountand--Assembly.mode2.phasing.maxDiscordantReadCount.
In addition, an edge is only generated if it carries sufficient useful information. For the simpler case of phasing, when bubbles in error have been removed, we can ignore the random hypothesis. We use the Bayesian model to compute
log(Pphasing) = | log [ Pposterior(in-phase) / Pposterior(out-of-phase)] |
Because of the absolute value, this gives the logarithmic spacing between the
in-phase and out-of-phase hypothesis.
A large value means that the edge strongly supports either the
in-phase or out-of-phase hypothesis.
The edge is only generated if
log(Pphasing)
is greater than the value specified
by command line option
--Assembly.mode2.phasing.minLogP.
This value specifies the minimum required value of
log(Pphasing), expressed in
decibels (dB).
The generated edge is then assigned a relative phase equal to 0 (in-phase) if
Pposterior(in-phase) > Pposterior(out-of-phase)
and 1 (out-of-phase) otherwise.
For bubble removal, things are a bit more complicated because now we also have to allow for the random hypothesis. For an edge to be generated we want it to decisively support either the in-phase or out-of-phase against both the random hypothesis and the opposite phasing hypothesis. Therefore in this case log(Pbubble-removal) for an edge is defined as follows:
If log [ Pposterior(in-phase) / Pposterior(out-of-phase) ] > 0 :
log(Pbubble-removal) = min { log [ Pposterior(in-phase) / Pposterior(out-of-phase) ] , log [ Pposterior(in-phase) / Pposterior(random) ] }
Otherwise:
log(Pbubble-removal) = min { log [ Pposterior(out-of-phase) / Pposterior(in-phase) ] , log [ Pposterior(out-of-phase) / Pposterior(random) ] }
In both cases, log(Pbubble-removal) is the logarithmic spacing between the best of the two phasing hypotheses against the next best hypothesis - either the opposite phasing hypothesis or the random hypothesis.
The edge is only generated if
log(Pbubble-removal)
is greater than the value specified
by command line option
--Assembly.mode2.bubbleRemoval.minLogP.
This value specifies the minimum required value of
log(Pbubble-removal), expressed in
decibels (dB).
Therefore the presence of an edge indicates good support for one of the phasing hypotheses, which in turn means that neither of the two bubbles are due to errors.
Bubble removal
Bubble removal is an iterative process. At each iteration we remove bubbles that don't phase well with adjacent bubbles. We create a phasing graph as described above and allowing the random hypothesis in the Bayesian model. This is done under control of the following command line options:
-
--Assembly.mode2.bubbleRemoval.minConcordantReadCount -
--Assembly.mode2.bubbleRemoval.maxDisconcordantReadCount -
--Assembly.mode2.bubbleRemoval.minLogP
- The phasing graph is created, with one vertex per bubble.
- Connected components are computed.
- All bubbles that correspond to vertices in small connected components
are flagged as bad and removed from the assembly graph
by only keeping the strongest branch.
A connected component is considered small if it contains
less than the number of bubbles specified via command line option
--Assembly.mode2.bubbleRemoval.componentSizeThreshold. - Any assembly graph simplifications made possible by the removal of some bubbles are made. This includes merging of adjacent linear segments and superbubble removal.
- The iteration continues by recreating the phasing graph from scratch, and stops when no more bubbles are flagged as bad.
Note this process can erroneusly flag as bad bubbles that are
in low heterozygosity regions of the genome.
This can cause low heterozygosity regions to be assembled haploid.
Mode 2 assembly works by phasing the bubbles, not the
reads. That is, the phasing process does not assign a
haplotype to reads. Rather, it assigns a haplotype to bubbles.
To achieve this,
the phasing graph is now created again,
but now the log(Pphasing)
for each edge is computed without allowing the random hypothesis,
as described
above.
We can do this because at this stage all bubbles in error
should have been removed.
Creation of the phasing graph is now under control of the
following command line options:
With the majority of bubbles in error having been removed,
the phasing graph is generally well behaved
and contains a small number of inconsistencies.
We assign to each edge a weight
equal to
log(Pphasing)
and compute an optimal spanning tree
that maximizes the sum of these weights.
With this definition, the spanning tree "prefers" edges
for which log(Pphasing) is large.
Once the spanning tree is computed, we assign
haplotypes arbitrarily on a randomly chosen bubble,
and then use the spanning tree to assign
haplotypes to the remaining bubbles.
This can be done using a Bread First Search (BFS) on the spanning tree
and assigning haplotypes to vertices based on the log(Pphasing)
values of the tree edges.
The spanning tree process has to be done
separately for each connected component
of the phasing graph, because there are no edges
the allow the BFS to jump across connected
components. This expresses the fact that
we cannot phase across regions in the absence of
a sufficient number of common reads.
The bubbles of each connected component
are then combined into a single vertex of the phasing graph,
keeping track of the relative phasing of the bubbles
as determined by the spanning tree.
This hierarchical phasing process is then repeated until all
vertices are isolated. Each vertex then corresponds to
a "phased component" in the final phased assembly.
At the end of this process, each bubble that was not
marked as bad has a
A bubble chain is a linear sequence
of diploid bubbles and intervening segments assembled haploid.
A portion of a phased bubble chain, as displayed in
Bandage,
is shown below.
On each diploid bubble, the two branches are colored
red or blue depending on the haplotype assigned
to each branch during phasing.
The intervening haploid segments are
shown in grey.
In most cases, there is a haploid segment
in between each pair of adjacent bubbles,
but sometimes two bubbles can immediately follow each other.
Bubble chains are useful for organizing assembly output.
As explained above, the phasing process assigns each
bubble to a connected component
and each branch to a haplotype.
With this information, we can find in the assembly graph paths
that represent each of the two haplotypes.
For example, in the above picture, we can
create a path that follows the haploid segments plus the
blue branches of diploid segments:
Paths created in this way provide haplotype separation on
a much longer scale than individual bubbles.
Haplotype separation can continue on both sides as long as we stay in
the same connected component.
A portion of a bubble chain in which all bubbles
belong
to the same connected component of the bubble graph
is called a phased region.
The intervening segments in between
adjacent phased regions on the same bubble chain are
called unphased regions.
The two paths for a phased region constructed as
defined above provide a representation of the
two haplotypes over the entire phased region.
This permits an assembly representation in which
each phased region is shown as a large
bubble, each of the two branches corresponding
to one of the two paths for the phased region.
After a mode 2 assembly run,
the resulting assembly is output in three different
representations containing different levels of detail:
All three representations also include the unstructured and unphased assembled segments
that are not part of a bubble chain.
For each of the three representations, four files are written out:
GFA files without sequence are much smaller
than the complete GFA files. So they can be useful to quickly
inspect the assembly in
Bandage
or to do other processing that does not require sequence.
FASTA files contain sequence in FASTA format
for all segments in each of the GFA files.
The csv files can be loaded in Bandage and used
to display information about the segments in each file.
They also define custom colors which facilitate
the display of the GFA files in Bandage.
Not all of the 12 files are necessary for all applications,
and so the following command line options can be used
to selectively suppress output:
Assembled segments in the detailed representation of the assembly
are named as follows:
Paths in the detailed representation of the assembly
are named like the corresponding segments in the phased representation
of the assembly and follow the segment naming schemes:
Bubble chains in the haploid representation of the assembly
are named
In the phased and haploid representations of the assembly,
segments that are not part of a bubble chain
have the same name as the corresponding segment
in the detailed representation of the assembly,
that is, a numeric identifier possibly followed by
Phased diploid assembly is turned on via
You can use `shasta --command listConfiguration --config
to get a ful description of each these two configurations,
including comments the describe in more details the conditions
under which eacxh of the two configurations were tested.
Please
file an issue
in the Shasta GitHub repository for discussion of mode 2 assembly parameters.
Assembly mode 2 requires the following setting (both
included in the configurations mentioned above):
Limited testing of phased diploid (mode 2) assembly was done
using current (Guppy 5) nanopore reads for a human genome.
Longer reads give better phasing results.
When using Ultra-Long (UL, N50>50Kb) reads
base-called with Guppy 5 and "super" accuracy
at coverage 60-80x, typical results for human genomes
are as follows:
Iterative assembly similarly addresses the separation of
copies of similar sequence due to ploidy, segmental duplication,
or other repeats.
It is more general in that it does not make
any assumption on the number of copies, while
assembly mode 2 has a built-in assumption of two copies.
However, iterative assembly uses a different approach:
it uses the assembly graph to separate reads and
then iteratively reassembles while enforcing
separation of the read pairs found in the previous
iteration to belong to different copies.
In summary, one could say that iterative assembly
"phases the reads", while mode 2 assembly
"phases the bubbles".
This approach used an iterative assembly, while effective in some cases, proved
to be much less robust and causes abundant assembly artifacts and
breaks. A future generalization of
mode 2 assembly without the assumption of two copies
may be able to provide robust separation
in the presence of arbitrary numbers of copies.
The Shasta assembler is designed to run on a single machine
with an amount of memory sufficient to hold all of its data structures
(1-2 TB for a human assembly, depending on coverage).
All data structures are memory mapped
and can be set up to remain available after assembly completes.
Note that using such a large memory machine does not substantially
increase the cost per CPU cycle. For example,
on Amazon AWS the cost per virtual processor hour for
large memory instances is no more than twice the cost for laptop-sized instances.
There are various advantages to running assemblies in this way:
To optimize performance in this setting, the Shasta assembler
uses various techniques:
Thanks to these techniques, Shasta achieves close to 100% CPU utilization
during its parallel phases, even when using large numbers of threads.
However, a number of sequential phases
remain, which typically result in average CPU utilization
during a large assembly around 70%. Some of these sequential
phases can be parallelized, which would result in increased
average CPU utilization and improved assembly performance.
As of Shasta release 0.1.0, a typical human assembly at coverage 60x
runs in about 6 hours on a
See here for information on
maximizing assembly performance.
--Assembly.mode2.phasing.minConcordantReadCount
--Assembly.mode2.phasing.maxDisconcordantReadCount
--Assembly.mode2.phasing.minLogP
componentId
that defines the final phasing graph vertex each bubble
was part of, and haplotypes
0 and 1 are assigned to its two branches.
Bubble chains
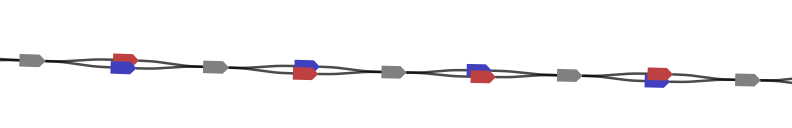
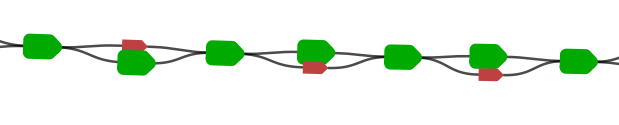
Mode 2 assembly output
All GFA files are in
GFA 1 format.
So in total, there are 12 output files, summarized in the table below:
Detailed
Phased
Haploid
Complete GFA file
Assembly-Detailed.gfa
Assembly-Phased.gfa
Assembly-Haploid.gfa
GFA file without sequence
Assembly-Detailed-NoSequence.gfa
Assembly-Phased-NoSequence.gfa
Assembly-Haploid-NoSequence.gfa
FASTA file
Assembly-Detailed.fasta
Assembly-Phased.fasta
Assembly-Haploid.fasta
FASTA file
Assembly-Detailed.csv
Assembly-Phased.csv
Assembly-Haploid.csv
--Assembly.mode2.suppressGfaOutput
turns off all GFA output.
--Assembly.mode2.suppressFastaOutput
turns off all FASTA output.
--Assembly.mode2.suppressDetailedOutput
turns off output of the Detailed representation of the assembly.
--Assembly.mode2.suppressPhasedOutput
turns off output of the Phased representation of the assembly.
--Assembly.mode2.suppressHaploidOutput
turns off output of the Haploid representation of the assembly.
Naming schemes for assembled segments and paths
12497.
.0 and .1.
For example, 17495.0 and 17495.1.
The portion preceding the period is the same for the
two branches of a bubble.
To clarify the above description, the figure below shows names for a portion
of bubble chain PR.bubbleChain.position.component.haplotype
(example: PR.17.41.268.1)
where PR stands for "Phased Region", and
the four fields are numeric identifiers with the following meaning:
bubbleChain identifies the bubble chain
that the segment belongs to.
position is the position of the segment in the bubble chain.
This begins at 0 and increments by one at each phased or unphased region
encountered in the bubble chain.
component identifies the connected component of the bubble
graph that this phased region belongs to.
haplotype (0 or 1) for each
of the two branches (haplotypes) of the phasing region. Usually a component
corresponds to a single phased region, but there are exceptions to that.
If two phased regions have the same component, their haplotypes are phased
relative to each other (that is, branch 0 of the first phased region
occurs in the same haplotype as branch 0 of the second phased region).
UR.bubbleChain.position
(example: PR.17.42)
where UR stands for "Unphased Region", and
the two fields are numeric identifiers with the same meaning
as for phased regions above.
0. Note how the position
field gets incremented along the bubble chain.
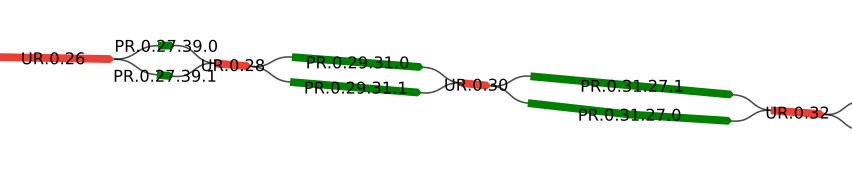
BC.bubbleChain where
bubbleChain identifies the bubble chain.
For example, BC.18.
.0 or .1.
Using mode 2 assembly
--Assembly.mode 2 or, more conveniently,
using command line option
--config
to specify one of the following configurations:
Nanopore-Phased-Jan2022.conf.
for standard nanopore reads.
Nanopore-UL-Phased-Jan2022.conf.
for Ultra-Long (UL) nanopore reads.
--Kmers.k must be even.
--ReadGraph.strandSeparationMethod 2
(strict strand separation in the read graph).
Assembly mode 2: typical results for diploid assembly
Comparison of assembly mode 2 and iterative assembly
High performance computing techniques
huge pages
). This makes it faster for
the operating system to allocate and manage the memory
and improves TLB efficiency. Using huge pages mapped on the
hugetlbfs filesystem
(Shasta executable options
--memoryMode filesystem --memoryBacking 2M)
can result in a significant speed up (20-30%) for large assemblies.
However, it requires root privilege via sudo.
Dynamic load balancing
). As a
thread finishes a piece of work assigned to it,
it grabs another chunk of work to do.
The process of assigning work items to threads is lock-free (that is, it uses atomic memory
primitives rather than mutexes or other synchronization methods provided by the operating system).
mmap
and can optionally be mapped to Linux 2 MB pages.
The Shasta code includes a C++ class for conveniently handling these large
memory-mapped regions as C++ containers with familiar semantics
(class shasta::MemoryMapped::Vector).
class shasta::MemoryMapped::VectorOfVectors). In the first
pass, one computes the length of each of the vectors. A single large area
is then allocated to hold all of the vectors contiguously,
together with another area to hold indexes pointing to the beginning of each of the short
vectors. In a second pass, the vectors are then filled. Both passes
can be performed in parallel and are entirely lock-free.
This process eliminates memory allocation overhead
that would be incurred if each of the vectors were to be
allocated individually.
x1.32xlarge AWS instance,
which has 64 cores (128 virtual CPUs) and 1952 GB of memory.